What if you could tap into the computational power of thousands of volunteers around the world to accelerate your drug discovery efforts? That’s exactly what we’ve done by integrating alchemiscale with Folding@Home, bringing “planetary scale” compute capacity to alchemical free energy calculations.
We recently presented this work at the 2025 Workshop on Free Energy Methods in Boston, and we’re excited to share it here.
the power of citizen science meets alchemical free energies#
Folding@Home is a remarkable citizen-science platform that harnesses the collective power of volunteers’ computers worldwide to run molecular dynamics simulations. During the COVID-19 pandemic, it became the first exascale computing system, reaching a staggering 2.43 exaflops of compute capacity. Among its many contributions, Folding@Home powered over 50,000 free energy calculations for the COVID Moonshot project, helping advance the search for a broadly-accessible COVID-19 antiviral.
Now in its 25th year of operation, Folding@Home continues to advance science across diverse applications—and we’re thrilled to bring this incredible resource to the OpenFE ecosystem through alchemiscale.
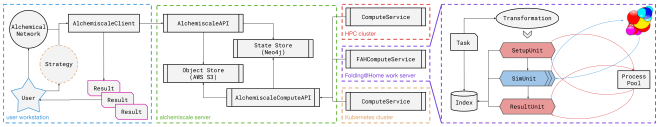
how it works: architecture for massive parallelism#
The integration required designing a specialized compute service that bridges alchemiscale’s orchestration capabilities with Folding@Home’s distributed execution model. We call this the FAHComputeService, implemented in alchemiscale-fah.
The key insight is task decomposition. When the FAHComputeService claims a free energy calculation task from alchemiscale, it breaks down the work into individual steps (called ProtocolUnits):
- Setup and analysis steps run locally via a process pool—these aren’t suitable for Folding@Home’s distributed execution model
- Compute-intensive simulation steps get dispatched to Folding@Home, where they’re distributed to volunteers worldwide
A single FAHComputeService can coordinate thousands of concurrent calculations, ensuring we keep Folding@Home well-fed with work whenever alchemiscale has tasks available. This architecture seamlessly extends alchemiscale’s existing support for HPC clusters, Kubernetes deployments, and individual workstations.
putting it to the test: 8,208 free energy calculations#
To validate the accuracy and performance of this approach, we ran a benchmarking study to using datasets from the recent OpenFE industry benchmark. Starting in April 2025, we prepared 21 alchemical networks spanning 4 public protein-ligand datasets (JACS, Fragments, MCS Docking, and Janssen BACE). For every ligand transformation, we ran identical calculations using the NonEquilibriumCyclingProtocol on both Folding@Home and conventional compute resources.
In summary, this comprised of:
- 8,208 total free energy calculations
- 4,104 on Folding@Home, 4,104 on conventional compute
- 1,012 ligand transformations completed successfully on both platforms
- roughly half of these ligand in solvent, half ligand in complex with receptor
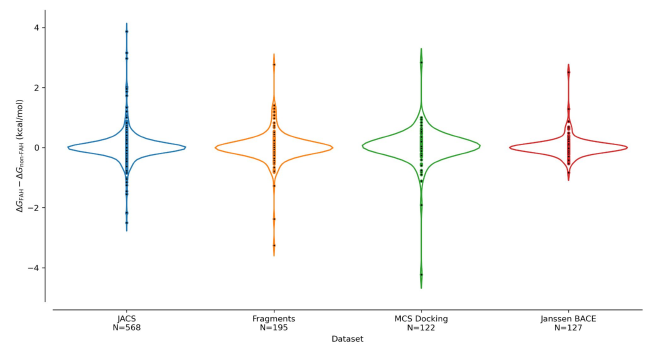
the results: accuracy meets scale#
accuracy: virtually indistinguishable#
Among the transformations that completed on both platforms, the differences between Folding@Home and conventional compute estimates gave:
- Mean difference: <0.04 kcal/mol
- Standard deviation: <0.60 kcal/mol
To put this in perspective, experimental binding affinity measurements typically have uncertainties around 0.5 kcal/mol. The Folding@Home results are statistically indistinguishable from conventional compute—exactly what we hoped to see.
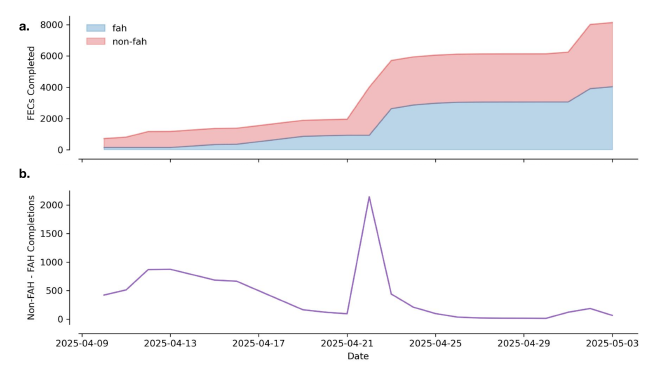
performance: trading latency for parallelism#
The performance story is more nuanced. Our approach to using Folding@Home does introduce additional latency compared to equivalently-provisioned conventional compute. This is expected given the distributed nature of the platform and the overhead of coordinating work across thousands of volunteer machines.
However, the trade-off becomes compelling under the right conditions. When conventional compute resources are under high contention—a common scenario in production environments—Folding@Home can deliver substantially more parallelism. During our benchmark, we observed over 2x the throughput of all our available conventional compute (comprised of two HPC clusters and one Kubernetes cluster).
The key is understanding when to leverage each resource:
- Conventional compute: Best when low latency is critical and resources are available
- Folding@Home: Ideal for large-scale campaigns where maximizing parallelism matters more than individual task turnaround time
what this means for the OpenFE ecosystem#
This integration opens exciting new possibilities:
Accelerated force field development: The ability to run thousands of validation calculations quickly will speed up the testing and refinement of new molecular mechanics force fields.
Large-scale open science: Folding@Home’s citizen-science model aligns perfectly with open science drug discovery initiatives. Projects with limited access to conventional compute can now tap into massive computational resources.
Benchmarking at scale: The OpenFE community can conduct more comprehensive benchmarks, testing methods across broader chemical spaces than previously practical.
Methods development: Researchers developing new approaches to network planning and atom mapping can validate their ideas with statistically robust sample sizes.
looking ahead#
The combination of alchemiscale’s orchestration capabilities and Folding@Home’s planetary-scale compute capacity represents a significant milestone for the OpenFE ecosystem. We’re continuing to refine the integration and explore new use cases, and are interested in developing additional alchemical protocols beyond non-equilibrium cycling for use on Folding@Home.
If you’re working on projects that could benefit from massive parallelism—whether that’s force field validation, large-scale drug discovery campaigns, or methods benchmarking—we’d love to hear from you. Join the conversation in our GitHub Discussions or reach out to the team.
A huge thank you to the Folding@Home team for their support in making this integration possible, and to the thousands of volunteers whose generosity makes this platform possible.
The full poster detailing this work is available here. This work was developed by datryllic, with funding and direction provided by ASAP Discovery and the Open Molecular Software Foundation.